
Contents
- 1 Mandatory arguments
- 2 Controlling results size
- 3 Sorting results by query IDs
- 4 Controlling speed
- 5 Optimizing quality/speed ratio using PLAST specific arguments
- 6 Optimizing PLAST: sample recipes
- 7 Optimizing PLAST at runtime: using seed-ratio
- 8 Optimizing PLAST at runtime: using threshold score
- 9 Optimizing PLAST at runtime: using max-database-size
- 10 Score matrix, gap costs and match/mismatch costs
- 11 Proteic-based PLAST search
- 12 Nucleic-based PLAST search (plastn only)
- 13 Monitoring job
Mandatory arguments
Each PLAST job requires at least to use the following arguments:
| Argument | Description | ||
|---|---|---|---|
| -p | comparison method. One of: plastn, plastp, plastx, tplastn or tplastx | ||
| -i | the query file provided as a Fasta formatted sequence file | ||
| -d | the reference databank. Either a Fasta file or a BLAST databank | ||
| -o | the results file |
As mentioned in the table, PLAST is capable of directly working with Fasta files. However, in the context of the reference databank, you can also provide a databank name when you have made use of KDMS (Korilog Databank Manager System) that is provided with KlastRunner. Such a databank name can be obtained by having a look at the KDMS graphical frontend: on the right panel, have a look at the column called “Name”.
Controlling results size
You can control how many hits are reported in a result file by using these arguments:
| Argument | Description | ||
|---|---|---|---|
| -e | E-value threshold. Default value is 10 | ||
| -max-hit-per-query | set the maximum number of hits aligned to a query. Default value is 10. Requires to use additional argument: -force-query-order 1000 |
||
| -max-hsp-per-hit | set the maximum number of HSPs reported for a hit. Default value is 1. Requires to use additional argument: -force-query-order 1000 |
If you want to get all possible hits/HSPs, simply pass 0 (zero) to -max-hit-per-query and -max-hsp-per-hit.
Sorting results by query IDs
Being a bank to bank sequence comparison tool, PLAST does not care about query ordering when producing results. It means that PLAST produces query/hit matches without any particular order. If you prefer PLAST producing results as BLAST, i.e. hits are sorted by query IDs, simply add the following argument to your PLAST command:
plast ... -force-query-order 1000 ...
Controlling speed
PLAST speed is of course controlled by the number of available cores:
| Argument | Description | ||
|---|---|---|---|
| -a | Number of cores. Default is the maximum number of cores available on the computer running PLAST. |
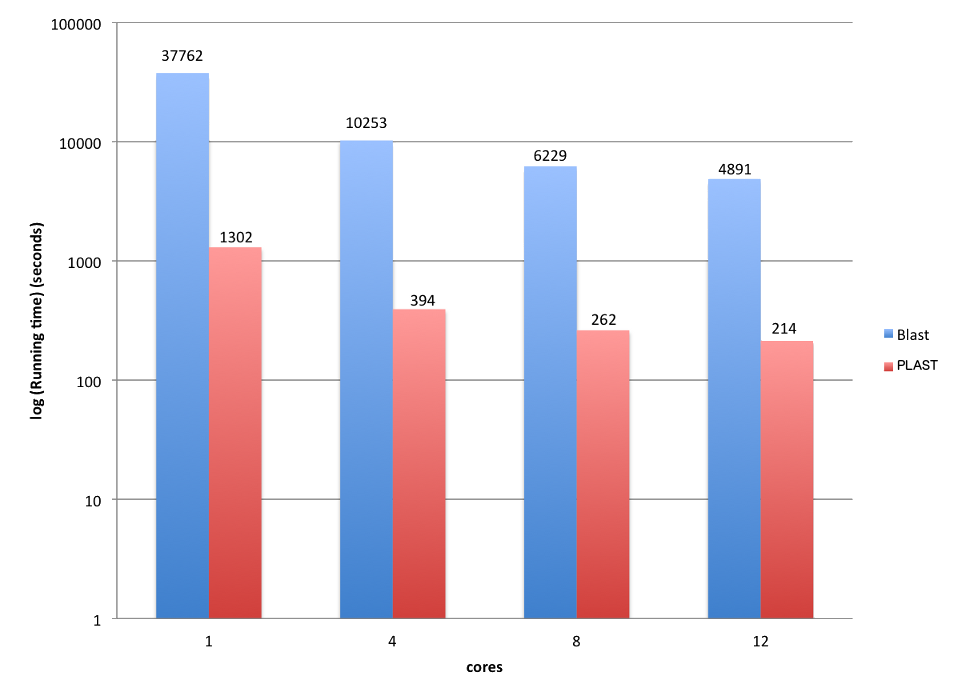
PLAST provides additional parameters to fine tune speed/quality ratio, as explained in the coming sections.
Optimizing quality/speed ratio using PLAST specific arguments
PLAST’s default configuration has been setup to provide an optimal ratio between speed and quality in order to produce results with quality similar to Blast. Even in such a configuration, you’ll have great speedup factors.
Depending on your needs you can enhance speed factors with little loss of quality in your results.
PLAST specific arguments for optimizing search jobs are:
| Argument | Description | ||
|---|---|---|---|
| -seeds-use-ratio | Ratio of seeds to be used (see comment, below). [1..100], default is 100. Decrease value to speedup algorithm with little loss of quality | ||
| -s | Ungapped threshold trigger a small gapped extension (see comment, below). [25..127], default is 38 and 55 for protein-based and nucleic-based comparisons, respectively. Increase value to speedup algorithm with little loss of quality. | ||
| -max-database-size | Maximum allowed size (in bytes) for a database. If greater, database is segmented (see comment, below) |
Fine tuning seed-ratio, threshold score and max-database-size may provide impressive acceleration of the KLAST comparison engine, with little loss of quality in the results. Carefuly read the following sections.
Optimizing PLAST: sample recipes
In order to tune PLAST correctly, we always invite our users to try the software with sample data sets. When you need to compare large set of sequences, always start your work by comparing a small subset of your data. This way, you can check the parameters, the results and the speed of the software.
As an example, if you have to compare 300,000 sequences against NCBI nt, start your work by comparing 300 query sequences against NCBI nt using default PLASTn parameters. Then, fine tune it (see below the use of seed-ratio, max-database-size and threshold score) and check the results. As soon as your parameters are fine, go ahead with 3,000 and/or 10,000 query sequences, and check results and speed. If everything is fine, then run the full comparison.
Optimizing PLAST at runtime: using seed-ratio
When using PLAST for protein-based sequence comparisons, the algorithm can be speedup using the seed-ratio parameter. As stated here, PLASTp algorithm relies on a finite table of seeds; there are about 6,200 seeds for BLOSUM50 and BLOSUM62 matrices, whatever the input sequence databanks (for more information, see Reference [1] here). During the comparison, PLAST orders seeds by occurrences, starting to process seeds producing the highest number of hits. So, it is possible to ask PLAST to use either the entire set of seeds to achieve a comparison, or a subset. This fine-tuning PLAST feature is achieved using the seed-ratio parameter, ranging from 1% to 100%. The highest seed-ratio you use, the highest sensitivity you get… the lowest seed-ratio you use, the highest speed you get with little loss in quality, as illustrated on this example:
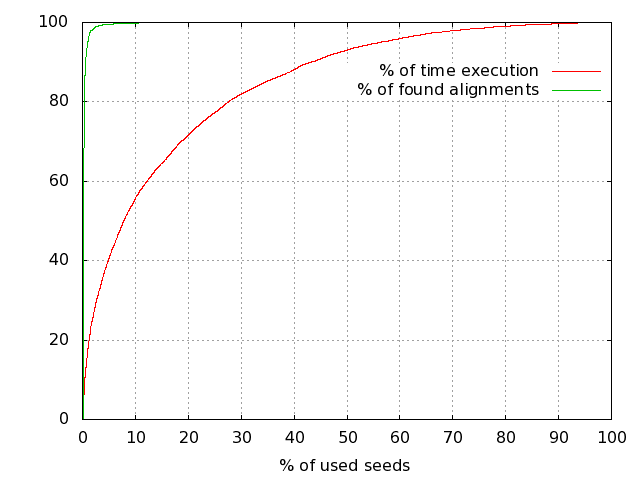
Using seed ratio to speedup Plast
Reducing number of seeds to use during a comparison still provides high results quality while dramatically reducing search time.
The seed-ratio parameter is available for PLASTp, PLASTx, tPLASTx and tPLASTn. When using PLAST from the command-line, use argument “-seeds-use-ratio value”, where “value” is in the range 1..100 (default is 100).
Optimizing PLAST at runtime: using threshold score
A second way to fine tune PLAST, and speedup the search, consists in using the ‘threshold score’ parameter. During a search, PLAST computes a score for each ungapped sequence alignment matching a query and a hit. As soon as this score is above the threshold, that alignment is retained for further processing. By default, this ‘threshold score’ (‘-s’ argument) is set to a small value (38 for protein comparisons, 55 for nucleotide comparisons) to let PLAST be as sensitive as possible. However, if you suspect that your query sequences may be closely related to the reference databank, you could increase the ‘threshold score’: PLAST can still produce high-quality results, but with an additional speedup.
Use case on 16s RNA
As an example, when comparing 900 reads (500 nucleotides on average) against Silva SSU databank (740,000 sequences) on a 8 cores Intel-Xeon based computer, search time was 73 hours using “-s 55”, but only 8 minutes using “-s 127”; results were the same in terms of quality, i.e. we got the same best hit for each query in both results.
Use case on protein and nucleotide comparisons
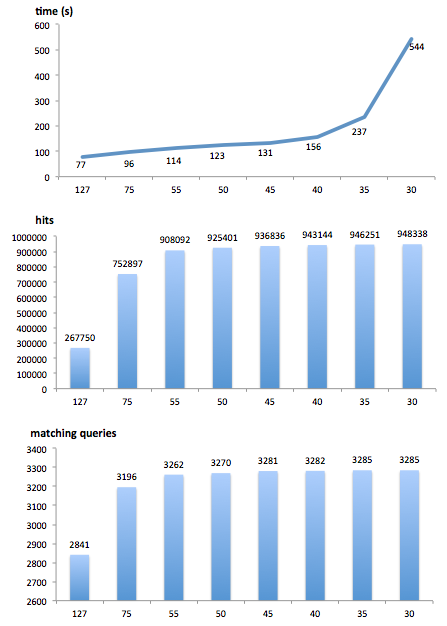
Use threshold score to speedup PLAST
Read these case studies about the optimal use of Threshold Score parameter
The threshold score parameter is available for PLASTn, PLASTp, PLASTx, tPLASTx and tPLASTn. When using PLAST from the command-line, use argument “-s value”, where “value” is in the range 25..127 (default is 38 and 55 for protein-based and nucleic-based comparisons, respectively).
Optimizing PLAST at runtime: using max-database-size
Another way to fine tune PLAST, and again speedup the search, consists in using the max-database-size parameter. It sets the amount of bytes to reserve in RAM in order to load databank pages into memory. Indeed, during the comparison of query vs. subject databanks, PLAST automatically paginates databanks if they do not fit entirely into RAM. For that purpose, PLAST relies on the max-database-size parameter; when setting up that parameter, compare the amount of RAM you have in your computer with ‘max-database-size x 8 x 2’ (each databank index requires ‘8 x max-database-size’ bytes, and you have two databanks). For instance, when using PLAST on a 32 Gb computer, increasing max-database-size from 20M (default value) to 100M may produce an additional speedup of 4x.
We advise you to setup the max-database-size value to enable the full load of the query databank into memory. For instance, if your query file sizes 12 Mb, then set the max-database-size parameter to 15000000 (15 Mb). Also, we do not recommend to set max-database-size to value above 100000000 (100Mb) ; in such a case, if your query file is very big, let PLAST paginates the query, or run several PLAST jobs, each of them processing one partition of your query file.
The max-database-size parameter is available for PLASTn, PLASTp, PLASTx, tPLASTx and tPLASTn. When using PLAST from the command-line, use argument “-max-database-size value”, where “value” is a number of bytes (default is 5000000).
Score matrix, gap costs and match/mismatch costs
PLAST arguments related to score matrix, gap costs and match/mismatch are presented in the following table.
| Argument | Description | ||
|---|---|---|---|
| -m | matrix | ||
| -G | cost to open a gap | ||
| -E | cost to extend a gap | ||
| -r | reward for a nucleotide match (plastn only) | ||
| -q | penalty for a nucleotide mismatch (plastn only) |
Argument -m (matrix) is only available for proteic-based comparison methods: plastp, plastx, tplastn and tplastx.
Arguments -r and -q (match/mismatch) are only available for plastn method.
Arguments -G and -E (gap costs) are available for all comparison methods: plastp, plastn, plastx, tplastn and tplastx.
Notice: in the following tables, (*) denotes the default value used by PLAST when you do not use a particular argument.
Proteic-based PLAST search
Valid score matrices and gap costs are as follows:
BLOSUM62
| Gap open | Gap extend | ||
|---|---|---|---|
| 11 | 2 | ||
| 11 | 1 | (*) | |
| 9 | 2 | ||
| 8 | 2 | ||
| 7 | 2 | ||
| 6 | 2 | ||
| 12 | 1 | ||
| 11 | 1 | ||
| 10 | 1 | ||
| 9 | 1 |
BLOSUM50
| Gap open | Gap extend | ||
|---|---|---|---|
| 13 | 3 | ||
| 13 | 2 | (*) | |
| 12 | 3 | ||
| 11 | 3 | ||
| 10 | 3 | ||
| 15 | 2 | ||
| 14 | 2 | ||
| 13 | 2 | ||
| 19 | 1 | ||
| 18 | 1 | ||
| 17 | 1 | ||
| 16 | 1 |
Nucleic-based PLAST search (plastn only)
Valid match/mismatch (arguments -r and -q) are as follows:
| Match | Mismatch | ||
|---|---|---|---|
| 1 | -1 | ||
| 1 | -2 | ||
| 1 | -3 | ||
| 1 | -4 | ||
| 2 | -3 | (*) | |
| 4 | -5 |
Given match/mismatch of 1,-1, valid gap costs are as follows:
| Gap open | Gap extend | ||
|---|---|---|---|
| 3 | 2 | (*) | |
| 2 | 2 | ||
| 1 | 2 | ||
| 0 | 2 | ||
| 4 | 1 | ||
| 3 | 1 | ||
| 2 | 1 |
Given match/mismatch of 1,-2, valid gap costs are as follows:
| Gap open | Gap extend | ||
|---|---|---|---|
| 5 | 2 | (*) | |
| 2 | 2 | ||
| 1 | 2 | ||
| 0 | 2 | ||
| 3 | 1 | ||
| 2 | 1 | ||
| 1 | 1 |
Given match/mismatch of 1,-3, valid gap costs are as follows:
| Gap open | Gap extend | ||
|---|---|---|---|
| 5 | 2 | (*) | |
| 2 | 2 | ||
| 1 | 2 | ||
| 0 | 2 | ||
| 2 | 1 | ||
| 1 | 1 |
Given match/mismatch of 1,-4, valid gap costs are as follows:
| Gap open | Gap extend | ||
|---|---|---|---|
| 5 | 2 | (*) | |
| 1 | 2 | ||
| 0 | 2 | ||
| 2 | 1 | ||
| 1 | 1 |
Given match/mismatch of 2,-3, valid gap costs are as follows:
| Gap open | Gap extend | ||
|---|---|---|---|
| 4 | 4 | ||
| 2 | 4 | ||
| 0 | 4 | ||
| 3 | 3 | ||
| 6 | 2 | ||
| 5 | 2 | (*) | |
| 4 | 2 | ||
| 2 | 2 |
Given match/mismatch of 4,-5, valid gap costs are as follows:
| Gap open | Gap extend | ||
|---|---|---|---|
| 12 | 8 | (*) | |
| 6 | 5 | ||
| 5 | 5 | ||
| 4 | 5 | ||
| 3 | 5 |
Monitoring job
PLAST enables you to monitor job execution:
plast ... -bargraph
When using argument -bargraph, PLAST displays a progression bar such as this one:
plastp [1/1] 100.0% align=16960 time [00:00:08 - 00:00:00 - 00:00:08] mem=298.7Mo (max=298.7Mo tot=0.3Go) seeds [5082:5082] [====================] 100%
Several pieces of information are provided, as follows:
| Element | Description |
| plastp | Name of comparison method |
| [1/1] | Pagination of reference databank. If reference databank fits in RAM at once, a single page is used to compare query and reference banks. |
| 100% | Progression of execution |
| align=16960 | Number of matches found |
| time […] | Three execution times are provided: ellapsed, remaining and total. It is worth noting that “remaining time” is always a predicted value. |
| mem (…) | Memory usages: currently used, maximum (peak) and cumulative (grand total over pagination of reference bank) |
| seeds […] | Number of seeds processed if the form [current seeds:total seeds] |
| [====….]x% | A text-based progression bar |