
PLAST relies on a new optimized and combined implementation of the Parallel Local Sequence Alignment Search Tool (PLAST) and the Ordered Index Seed (ORIS) algorithms developed by Inria’s Genscale research team, headed by Dominique Lavenier, pioneer in hardware and software accelerators dedicated to large scale genomics applications.
The first part of this article gives you the major features of the PLAST algorithm (i.e. how it works). Then, the second part will give you some practical ways to fine tune the method in order to find a very good balance between high-speed and results quality.
Contents
PLAST algorithm presentation
PLAST aims at comparing two large sets of sequences. Query and subject can be both proteins or nucleotides for which you’ll use PLASTp or PLASTn to compare them, respectively. Considering proteins versus translated nucleotides, you’ll use one of PLASTx, tPLASTx or tPLASTn. PLASTp, PLASTx, tPLASTn and tPLASTx algorithms are actually using the same algorithm, relying on PLASTp principles. PLASTn uses a slightly different approach. The following sections describes PLASTp and PLASTn.
Protein-protein comparison (plastp and “plastp-like”: plastx, tplastn, tplastx)
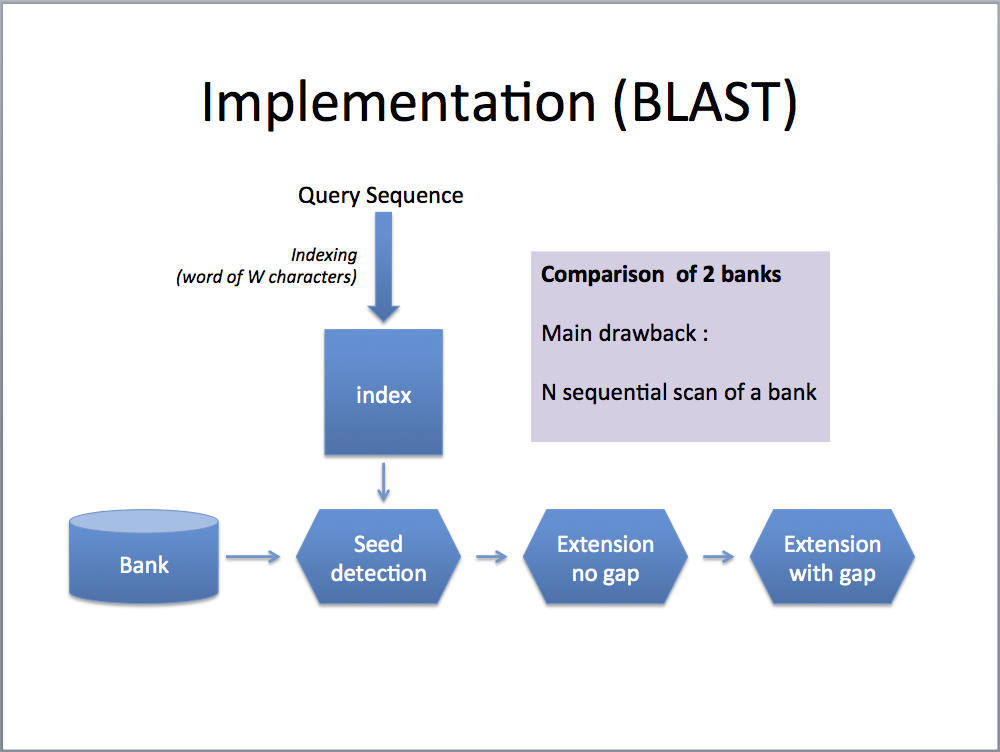
PLASTp aims to compare two large sets of sequences. It implements a three-step seed-based algorithm: (1) indexing, (2) ungap extension and (3) gap extension. Compared to BLASTp algorithm, PLASTp differs essentially during step 1. Steps 2 and 3 are similar between the two algorithms.
- Both sets are first indexed with subset-seeds of W characters [1]. The index is a table of K entries (K is the number of different seeds). Each entry is associated with a list L of P positions corresponding to all occurrences of the seeds in the sequence set.
- For each seed, P1 x P2 ungap extensions are performed. P1 (resp. P2) represents the number of elements associated to the list L1 (resp. L2) for the selected seed. Ungap extensions are computed on subsequences made of 2L+W characters (size of the seed + L neighboring residues before and after the seed). Ungap alignments with a score greater than a predefined threshold value are selected for the 3rd step.
- As the previous step can generate many ungap alignments belonging to the same gap alignment, a checking on the final set of alignments is done before launching the gap process. Dynamic programming algorithm is used to compute final gapped sequence alignments.
Moreover, PLASTp is optimized for multicore processor architectures. The algorithm is natively parallelized following a coarse-grained approach scaling well with the number of cores. Furthermore, gap and ungap extension computation are vectorized using the SSE instruction set of processors.
Nucleotide-nucleotide comparison (plastn)
PLASTn compares two large sets of DNA sequences. Similarly to PLASTp, two indexes including neighboring information are built, allowing the first steps of the search to be fastened. Furthermore, PLASTn includes a new search engines based on the concept of Ordered Seed Indexing developed by D. Lavenier [3]. This algorithm suppresses numerous redundant computations during the extension step of alignment construction.
As PLASTp, PLASTn is optimized for today multicore microprocessor architectures, and scale well with the number of cores.
Differences between PLAST and BLAST algorithms
BLAST and PLAST almost use the same heuristic to compare sequences: they use seeds, they create optimal gapped-alignment with Smith-Waterman algorithm and they use the same statistical model to compute scores and e-values and they provide all comparison methods (protein vs. protein, nucleotide vs. nucleotide, translated nucletoide vs. protein, etc.).
However, PLAST is fully designed to compare two set of sequences, i.e. the query is made of many sequences. Of course, you can use PLAST to compare a single query sequence vs. a subject databank, but you won’t get any speedup in comparison to BLAST. Another difference between PLAST and BLAST remains in the software implementation: PLAST extensively uses processor cache memory, SIMD instructions set (SSE3), multi-threading and double-indexing scheme.
 |
 |
References
[1] Roytberg M, Gambin A, Noe L, Lasota S, Furletova E, Szczurek E, Kucherov G: On subset seeds for protein alignment. IEEE/ACM Trans Comput Biol Bioinformatics 2009, 6(3): 483-494. Link.
[2] Nguyen VH, Lavenier D: PLAST: parallel local alignment search tool for database comparison. BMC Bioinformatics 2009, 10:329. Link.
[3] Lavenier D., Ordered Index Seed Algorithm for Intensive DNA Sequence Comparison, HiComb 2008: IEEE International Workshop on High Performance Computational Biology, Miami, Florida, 2008. Link.